генетична консультація
Генетика – область біології, яка найбільш динамічно розвивається і в даний час впливає на кожну галузь медицини, вона змінила наш підхід до походження хвороб, їх лікування та профілактики.

Пересічна людина пов’язує її з вродженими вадами, з декількома синдромами розумової відсталості, останнім часом з раком. Але генетики вже знають, що в кожній відомій хворобі вона бере безпосередню чи опосередковану участь. Часто поширений діабет, атеросклероз, згустки крові, закупорки, інфаркти, аритмії або психіатричні синдроми – там всюди можна знайти генетичну основу.
Генетика – це нова клінічна галузь, надзвичайно складна, мультидисциплінарна, і, можливо, саме тому в Польщі зараз трохи більше 100 клінічних генетиків.
У даний час відомо понад 23 000 захворювань та генетичних синдромів, і щодня в індексі OMIM з’являються нові. У більшості випадків відома їх молекулярна основа, але все ще існує значна кількість нелокалізованих мутацій. Тисячі молекулярних біологів та працівників лабораторій в інститутах по всьому світу шукають розташування генів, відповідальних за хворобу.
ДНК людини має довжину близько 2 метрів і містить мільйони генів, тому сам факт прочитання геному людини ще не дає відповіді на всі питання, де саме розташовані гени, що відповідають за функції органів, найскладнішим з яких є мозок людини.
Ці 2 метри ДНК правильно складені та упаковані у вигляді хромосом у кожній клітині. У кожній хромосомі є так звані активні місця (еухроматин) і уявно неактивні (гетерохроматин). «Уявно», оскільки їх біологічна функція на сьогодні чітко не доведена, але вони точно її мають. При деяких захворюваннях довжина їх специфічних послідовностей або кількість повторів у безпосередній близькості від гена впливає на функцію гена, хоча він побудований правильно. Однією з таких серйозних хвороб є хвороба Гантінгтона, важка смертельна хвороба, яка з’являється лише в зрілому віці. Дуже подібний механізм виступає при так званій сімейній розумовій відсталості, тобто синдромі FraX, який зустрічається переважно у хлопчиків, але передається матір’ю. Таких прикладів багато.
Отже, найдавніші докази зв’язку між хворобами та генетичними розладами походили від того, що можна було побачити під мікроскопом, від цілих хромосом. Відомі синдроми: Дауна, Едвардса, Патау або Тернера і Клайнфельтера – це порушення кількості цілих хромосом – як їх надлишку, так і відсутності. Оскільки у природі кількість=якість, тому як відсутність всієї хромосоми, так і її надлишок викликають порушення в однаковій мірі. У найпоширенішій у світі трисомії – синдромі Дауна виступає додаткова ціла хромосома 21 пари, і цей надлишок порушує роботу двох інших хромосом, що призводить до загальновідомих симптомів. Це один з найслабших перебігів трисомії. Інші трисомії, наприклад хромосоми 1, 9 або 16, настільки важкі, що самостійно викликають викидень зазвичай уже в першому триместрі вагітності, а інші трисомії, наприклад 13 і 18 пар, є летальними (смертельними) до або незабаром після пологів (рис. 1).
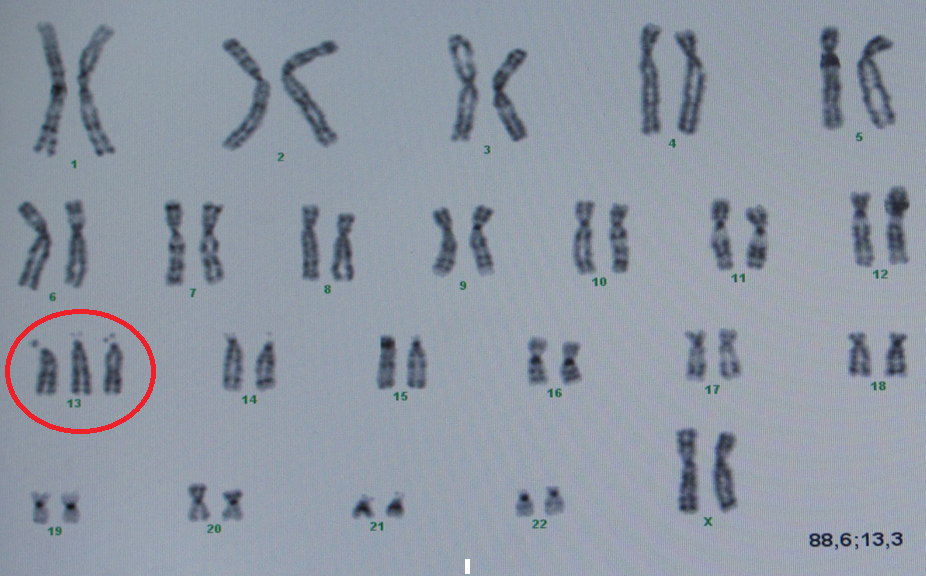
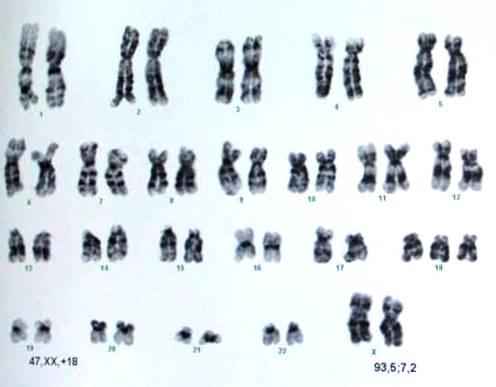
Рис. 1. Надлишок хромосом при синдромі Патау (трисомія 13), важкому, смертельному захворюванні.
Рис. 2. Надлишок хромосом при синдромі Едвардса (трисомія 18) – смертельному захворюванні
Однак не тільки вся зайва хромосома, але навіть невелика частина її надлишку також спровокує повномасштабний синдром Дауна. Існують трисомії 21 пари, так звані транслокаційні, які можна успадкувати з дуже великою ймовірністю. На щастя, таких випадків небагато. Наведений вище простий приклад дозволяє усвідомити, що при такій добре відомій хворобі можуть існувати різні її варіанти, переважно не спадкові (регулярні), але також передані батьками.
У кожній клітині є певна кількість відділів, і в кожному з них природа може помилитись. Однак у клітині є механізми репарації, але й вони можуть підвести з різних причин. Якщо помилка виникає в яйцеклітині або сперматозоїді, то плід уражається хворобою від одноклітинної форми. Нерепаровані мутації також трапляються в ембріональному, внутрішньоутробному та дорослому житті. Специфічним «охоронцем геному» є білок р53, закодований у гені на хромосомах 17 пари. Мутації цього гена виявляються приблизно в половині всіх видів раку, які також є наслідком порушення функції репарації ДНК.
Не тільки патологічна кількість хромосом викликає захворювання. Втрати або надлишки хромосомних мікрофрагментів, які звуться мікроделеціями або дуплікаціями, є однаково вагомими. Парадоксально, але ці розлади дуже поширені і викликають надзвичайно серйозні захворювання. Думаю, багато Матерів воліють виховувати дитину з синдромом Дауна, ніж з однією з мікроделецій. Тяжкість цих дефектів обумовлена повною втратою функції тисяч відсутніх генів, а їх локалізація викликає специфічні симптоми (рис. 3).
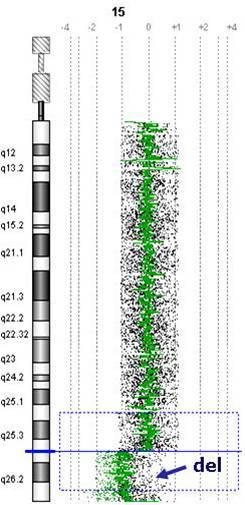
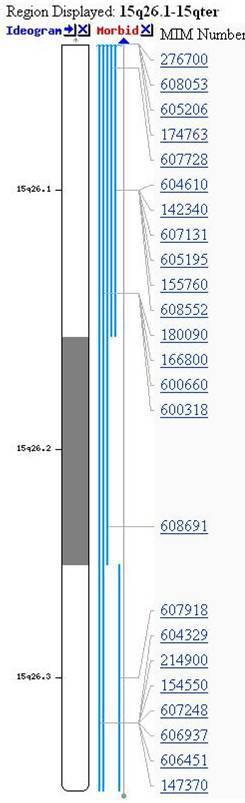
Рис. 3. Мікроделеція в хромосомі 15 та поруч найважливіші гени, які через це порушення відсутні в одній копії генів, що є причиною повної або часткової відсутності їх функції та в результаті важких дефектів та порушень функції організму.
Наступною групою розладів є так звані моногенні захворювання. Кожен ген складається з сотень або тисяч пар кодів. Парадоксально, але досить помилитися лише одним кодом і замінити його іншим – неправильним, щоб ген не працював належним чином і виробляв неправильний білок. Наслідки такої помилки можуть бути катастрофічними і спричиняють одну з найважчих груп захворювань. Приклади включають муковісцидоз (Кістозний фіброз) або метаболічні захворювання. Інші фактори також можуть порушити функцію генів, не лише відсутність належного коду. Це може бути аномальний зразок метилювання (СН3) у гені, або, наприклад, активність неправильного гена в його парі (материнського чи батьківського) та багато інших аномалій.
Окреме питання – це питання спадковості хвороб, оскільки «генетична хвороба» не завжди дорівнює «спадковій хворобі». Навіть дуже важкі розлади можуть трапитися «випадково» і не повинні бути спадковими, як це буває при трисоміях з надлишком цілої хромосоми в даній парі, наприклад, найпоширеніших, так званих регулярних трисоміях 13, 18 та 21. Деякі хвороби можуть успадковуватися аутосомно-домінантним (AD) способом, який означає, що якщо один із батьків має захворювання, симптоми ЗАВЖДИ з’являться у їхніх нащадків. Багато захворювань успадковуються за допомогою аутосомно-рецесивного (AR) способу, що означає, що дитина повинна успадкувати дефектні гени від обох батьків, щоб у неї з’явилися симптоми. Це дуже підступні і зазвичай серйозні розлади, наприклад, при муковісцидозі, але батьки можуть бути лише носіями дефектного гена.
Інші типи успадкування це, наприклад, пов’язані з Х-хромосомою, безсимптомним носієм захворювання є тоді мати, а частіше за все хворіють діти чоловічої статі. Ми знаємо ще багато інших моделей успадкування, але це виходить за рамки скороченої інформації.
Клінічний генетик, спираючись на племінні, клінічні, цитогенетичні та молекулярні дані, завжди визначає так звану ймовірність виникнення даного захворювання у нащадків чи інших членів сім’ї, і часто ситуація не є такою простою, як може здатися. Від найпростішої моделі 50%/50% при захворюваннях AD, через не менш поширені 25%/75% при захворюваннях АR, до складних моделей в пропорційних транслокаціях, полігенних захворювань та генетики популяцій.
Подібна «ймовірність» застосовується також у пренатальній діагностиці, при якій визначається ризик виникнення, наприклад, найпоширеніших трисомій, однак у цих випадках враховується дуже багато різних факторів: крім даних родоводу, включаючи вік матері, вагу, тижні вагітності, кількість білка, що циркулює в її крові, аж до численних так званих ультразвукових маркерів. Однак у цьому дослідженні необхідні розширені комп’ютерні програми, які пов’язують між собою десятки отриманих даних, здавалося б, не пов’язаних між собою. Однак пренатальна генетика є набагато заплутанішою та складнішою сферою. Під час обстеження дитини чи дорослого ми зазвичай маємо інший, практично необмежений часовий масштаб. У пренатальній медицині час відіграє ключову роль, результат потрібно отримати набагато швидше, важливими є тижні, часто навіть дні! Матеріал для тестування також є набагато складнішою речовиною, оскільки, наприклад, при типовому амніоцентезі його дуже мало. Поодинокі досліджувані клітини плоду, як правило, походять із відшарованого епітелію плоду або сечі плоду, їх мало, вони дуже погано вирощуються, і виділення відповідної кількості ДНК із такого дефіцитного матеріалу вдається лише деяким особам і вимагає спеціальних навичок. У пренатальній медицині ми також частіше не можемо поставити діагноз, особливо при рідкісних метаболічних захворюваннях, при яких симптоми проявляються дуже пізно (наприклад, дефекти кісток) або лише в новонародженій чи дитячій стадії. Слід також пам’ятати, що генетика дає лише відповідь ТАК чи НІ на задане питання щодо конкретного захворювання. Передбачення такого захворювання, особливо рідкісного, вимагає спеціальних знань та навичок, тому акушери та генетики завжди співпрацюють між собою у всіх авторитетних пренатальних лабораторіях. Тільки таким чином можна досягти пренатального успіху, що безпосередньо перетворюється на швидкий і точний діагноз, що забезпечує спокій і обізнаність матері та можливість допомогти дитині.
Доктор Медичних Наук Кшиштоф Піотровський
Спеціаліст з Клінічної Генетики
REJESTRACJA
Godziny otwarcia
pon.- cz. 8:00-20:00