Poradnictwo genetyczne

Przeciętnemu człowiekowi kojarzy się z wadami wrodzonymi, z kilkoma zespołami upośledzenia umysłowego, ostatnio z nowotworami. Genetycy wiedzą już jednak, że w każdą znaną chorobę jest uwikłana bezpośrednio lub pośrednio. Powszechnie występująca cukrzyca, miażdżyca, zakrzepy, zatory, zawały, zaburzenia rytmu czy zespoły psychiatryczne – wszędzie tam można znaleźć podłoże genetyczne.
Genetyka jest nową dziedziną kliniczną, wyjątkowo trudną, wielospecjalistyczną i być może dlatego właśnie, genetyków jest mało – w Polsce obecnie niewielu ponad 100, jednak są kraje, gdzie w każdym szpitalu musi być zatrudniony genetyk, co potwierdza jej rosnące znaczenie.
Obecnie znanych jest ponad 23 tysiące chorób i zespołów genetycznych i każdego dnia w indeksie OMIM pojawiają się nowe. W większości przypadków poznano ich podłoże genetyczne, jednak nadal pozostaje znaczna liczba o niezlokalizowanych mutacjach. Poszukiwaniem lokalizacji genów odpowiedzialnych za chorobę zajmują się tysiące biologów molekularnych i laborantów w instytutach na całym świecie. Ponieważ wzór DNA jest uniwersalny u wszystkich istot żywych, przeto wielką pomocą są badania na innych formach żywych od muszki owocowej począwszy, na pewnych gatunkach małp kończąc.
Długość ludzkiego DNA wynosi około 2 metry, a w jego składzie znajdują się miliony genów, zatem sam fakt odczytania ludzkiego genomu nie dał jeszcze odpowiedzi na wszystkie pytania, w których jego miejscach, zlokalizowane są geny odpowiedzialne za funkcje narządów, z których najbardziej skomplikowany jest ludzki mózg.
Owe 2 metry DNA są w każdej komórce odpowiednio zwinięte i upakowane w postaci chromosomów. W każdym chromosomie są tzw. miejsca aktywne (eurochromatyna) i rzekomo nieaktywne (heterochromatyna). „Rzekomo”, ponieważ dotychczas jasno nie udowodniono ich funkcji biologicznej, jednak z pewnością takową posiadają. W niektórych chorobach długość ich specyficznych sekwencji lub liczba powtórzeń w bezpośrednim sąsiedztwie genu, wpływa na funkcję genu, pomimo, iż jest on zbudowany prawidłowo. Jedną z takich ciężkich chorób jest pląsawica Huntingtona, ciężka śmiertelna choroba, która ujawnia się dopiero w wieku dojrzałym. Bardzo podobny mechanizm występuje w rodzinnym upośledzeniu umysłowym tzw. Zespole FraX, który występuje głównie u chłopców, jednak przenoszony jest przez matkę. Przykłady można mnożyć.
Najstarsze dowody na powiązania chorób z zaburzeniami genetycznymi pochodziły zatem od tego, co można było zauważyć w mikroskopie, od całych chromosomów. Znane zespoły: Downa, Edwardsa, Patau’a czy Turnera i Klinefeltera – to zaburzenia liczby całych chromosomów – zarówno ich nadmiaru, jak i braku. W naturze bowiem ilość = jakość, zatem zarówno brak całego chromosomu, jak i jego nadmiar w równym stopniu wywołuje zaburzenie. W najczęstszej w populacji na świecie trisomii – Zespole Downa, występuje dodatkowy cały chromosom 21 pary i ten nadmiar zaburza pracę pozostałych dwóch chromosomów, prowadząc do znanych powszechnie objawów. Jest to jedna z najłagodniej przebiegających trisomii. Inne trisomie, np. chromosomu 1, 9 czy 16 są tak ciężkie, że ulegają samoistnemu poronieniu zazwyczaj już w I trymestrze ciąży, a jeszcze inne trisomie np.: 13 i 18 pary są letalne (śmiertelne) przed lub w niedługim czasie po porodzie (ryc.1,2).
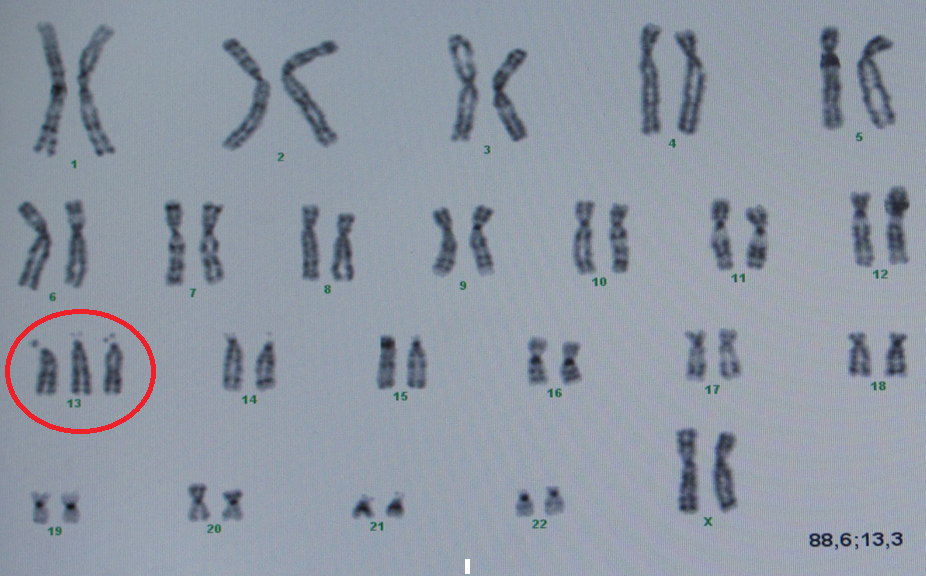
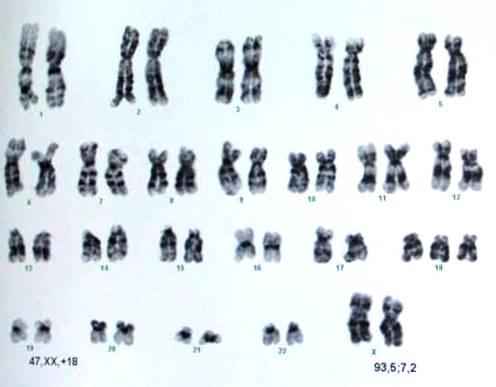
Jednak nie tylko cały dodatkowy chromosom, ale nawet drobny fragment jego nadmiaru wywoła także pełnoobjawowy zespół Downa. Istnieją również trisomie 21 pary tzw. translokacyjne, które mogą się dziedziczyć i to z bardzo dużym prawdopodobieństwem. Szczęśliwie takich przypadków jest mało. Powyższy prosty przykład pozwala uzmysłowić sobie, iż w tak dobrze poznanej chorobie, mogą być różne jej warianty, w większości niedziedziczne(regularne), jednak także przenoszone przez rodziców.
W każdej komórce zachodzi określona liczba podziałów i podczas każdego z nich , natura może się pomylić. W komórce są jednak mechanizmy naprawcze, lecz i one mogą zawodzić z różnych przyczyn. Jeśli pomyłka nastąpi w komórce jajowej lub plemniku, wówczas płód, już od formy jednokomórkowej jest dotknięty chorobą. Mutacje nie naprawione zachodzą także w życiu embrionalnym, płodowym i dorosłym. Swoistym „strażnikiem genomu” jest białko p53 zakodowane w genie na chromosomach 17 pary. Mutacje w tym genie odnajduje się w około połowie wszystkich nowotworów, które są także wynikiem zaburzeń funkcji naprawczych DNA.
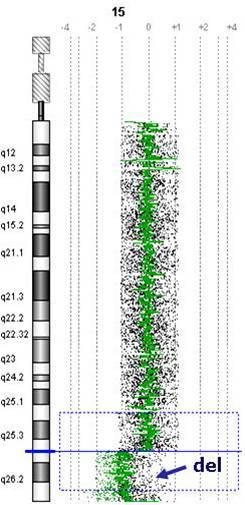
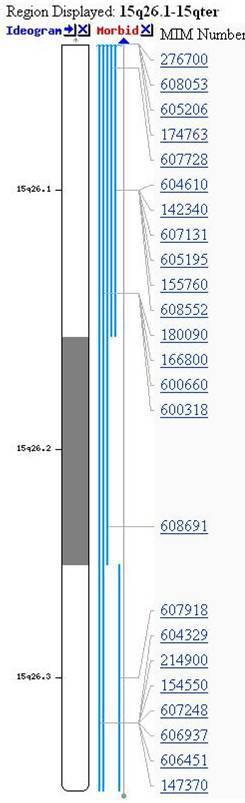
Nie tylko zaburzona liczba chromosomów powoduje choroby. Równie ciężkie są ubytki lub naddatki mikrofragmentów chromosomowych nazywane mikrodelecjami bądź duplikacjami. Te zaburzenia paradoksalnie występują bardzo często i są przyczyną wyjątkowo ciężkich chorób. Myślę, iż niejedna Matka wolałby wychowywać dziecko z Zespołem Downa, aniżeli z którąś z mikrodelecji. Ciężkość tych defektów wynika z całkowitego wypadnięcia funkcji tysięcy brakujących genów, a ich lokalizacja powoduje określone objawy (ryc.3.).
Następną grupą zaburzeń są tzw. choroby jednogenowe. Każdy gen składa się z setek lub tysięcy par zasad. Paradoksalnie wystarczy pomyłka jednej tylko zasady i zastąpienie jej inną – niewłaściwą, aby gen nie funkcjonował prawidłowo i produkował nieodpowiednie białko. Skutki takiej pomyłki bywają katastrofalne i wywołują jedne z najcięższych grup chorób. Przykładami może być mukowiscydoza (Cystic Fibrosis) czy choroby metaboliczne. Funkcję genów mogą zaburzyć także inne czynniki, nie tylko brak właściwej zasady. Może to być nieprawidłowy wzór metyzacji (CH3 ) w genie lub np. aktywność niewłaściwego genu z jego pary (matczynego lub ojcowskiego pochodzenia) i wiele wiele innych nieprawidłowości.
Odrębną kwestią jest kwestia dziedziczności chorób, bowiem „choroba genetyczna” nie zawsze równa się „chorobie dziedzicznej”. Nawet bardzo ciężkie zaburzenia mogą wydarzyć się „przypadkowo” i nie muszą mieć charakteru dziedzicznego, jak to ma miejsce w trisomiach z nadmiarem całego chromosomu w danej parze np.: najczęstsze tzw. regularne trisomie 13, 18 i 21. Część chorób może być dziedziczona w sposób autosomalnie dominujący (AD) co oznacza, iż w przypadku choroby u jednego z rodziców objawy ZAWSZE wystąpią u potomstwa. Wiele chorób dziedziczy się w sposób autosomalnie recesywny (AR) co oznacza, iż dziecko musi odziedziczyć wadliwe geny od obydwojga rodziców, aby u niego wystąpiły objawy. Są to bardzo zdradliwe i zazwyczaj ciężkie zaburzenia np. w mukowiscydozie, jednak rodzice mogą być jedynie nosicielami wadliwego genu.
Inne typy dziedziczenia to np.: sprzężone z chromosomem X, bezobjawowym nosicielem choroby jest wówczas matka, a chorują najczęściej dzieci płci męskiej. Znamy jeszcze wiele innych modeli dziedziczenia, jednak wykracza to poza ramy skrótowych informacji.
Lekarz genetyk kliniczny, na podstawie danych rodowodowych, klinicznych, cytogenetycznych i molekularnych określa zawsze tzw. prawdopodobieństwo wystąpienia danej choroby u potomstwa lub innych członków rodziny i często sytuacja nie jest tak prosta, jak pozornie mogłoby się wydawać. Od najprostszego modelu 50%/50% w chorobach AD, przez równie częste 25%/75% w chorobach AR, aż do skomplikowanych wzorów w translokacjach zrównoważonych, chorobach poligenowych i genetyce populacji.
Podobnym „prawdopodobieństwem” operuje także diagnostyka prenatalna, w której określa się ryzyko wystąpienia np. najczęstszych trisomi, jednak w tych przypadkach bierze się pod uwagę bardzo liczne i różnorodne czynniki: oprócz danych rodowodowych, łącznie z wiekiem matki, wagą, tygodniami ciąży, wartościami białek krążących w jej krwi, aż po liczne tzw. markery ultrasonograficzne. W tych jednak badaniach niezbędne są rozbudowane programy komputerowe, sprzęgające ze sobą dziesiątki uzyskanych danych, pozornie nie powiązanych ze sobą. Genetyka prenatalna jest jednak dziedziną znacznie trudniejszą i skomplikowaną. Badając dziecko lub dorosłego dysponujemy zazwyczaj inną skalą czasową, właściwie nieograniczoną. W medycynie prenatalnej czas gra kluczową rolę, wynik musimy uzyskać znacznie szybciej, rolę grają tygodnie, niejednokrotnie dni! Także materiał do badania jest znacznie trudniejszym tworzywem, bowiem np. w typowej amniopunkcji jest on bardzo skąpy. Pojedyncze, badane komórki płodu pochodzą zazwyczaj ze złuszczonego nabłonka płodu lub moczu płodu, jest ich niewiele, bardzo źle się je hoduje, a wyizolowanie z tak skąpego materiału odpowiedniej ilości DNA dane jest tylko nielicznym jednostkom i wymaga szczególnych umiejętności. W medycynie prenatalnej częściej także nie jesteśmy w stanie postawić diagnozy, zwłaszcza w chorobach rzadkich, metabolicznych, w których objawy pojawiają się bardzo późno (np.:wady kości) lub dopiero w okresie noworodkowym lub dziecięcym. Należy także pamiętać, iż genetyka daje jedynie odpowiedź na zadane pytanie w zakresie TAK lub NIE w stosunku do określonej choroby. Wytypowanie takiej choroby, zwłaszcza rzadkiej, wymaga szczególnej wiedzy i umiejętności, dlatego we wszystkich renomowanych pracowniach prenatalnych współpracują ze sobą zawsze na bieżąco położnicy i genetycy. Tylko w ten sposób można osiągnąć sukces prenatalny, który bezpośrednio przekłada się na szybką i trafną diagnozę, zapewniając spokój i wiedzę dla matki i ewentualną możliwość pomocy dziecku.
dr n. med. Krzysztof Piotrowski
Specjalista Genetyki Klinicznej
REJESTRACJA
Godziny otwarcia
pon.- cz. 8:00-20:00